Driving treatment decisions on 10 Nov 2015
Scientists use cancer models understand cancer. These models describe the rules cancer follows – how it starts, grows, metastasizes, and ultimately how it can be killed. Reviews of models generate new treatment ideas that models indicate should be successful.
Models of how cancer works drive the direction of cancer funding, research, prevention and treatment decisions. Faulty models lead to research producing ineffective treatments. Some researchers say that is happening now.
This post introduces the cancer stem cell model. The cancer stem cell model is not widely accepted but is gaining traction and a share of research money. It was developed as an alternative to the clonal evolution model to explain treatment failures.
Cancer Stem Cell Model
The cancer stem cell (CSC) model is an alternate model to explain the origin and diversity of cancer – and why past treatments have failed2. This model says that some (perhaps all) cancers are driven by a small number of treatment-resistant cancer cells with stem cell-like properties3,5. Stem cells have a slower life cycle and thus are largely unaffected by traditional chemotherapies that disrupt rapidly-dividing cells.6
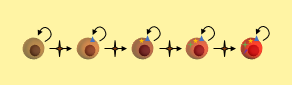
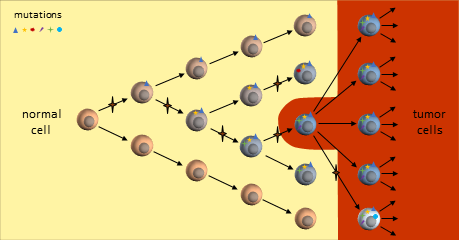
Cancer Stem Cells Accumulate Mutations
[The Cancer Stem Cell model was hard to research. There are many papers and web seminars that present their research as, “this is the way it is – no disputes”. Sorting out what is commonly accepted or not takes a lot of review and I’m certain I don’t have it all correct. This is the best I’ve come up with.]
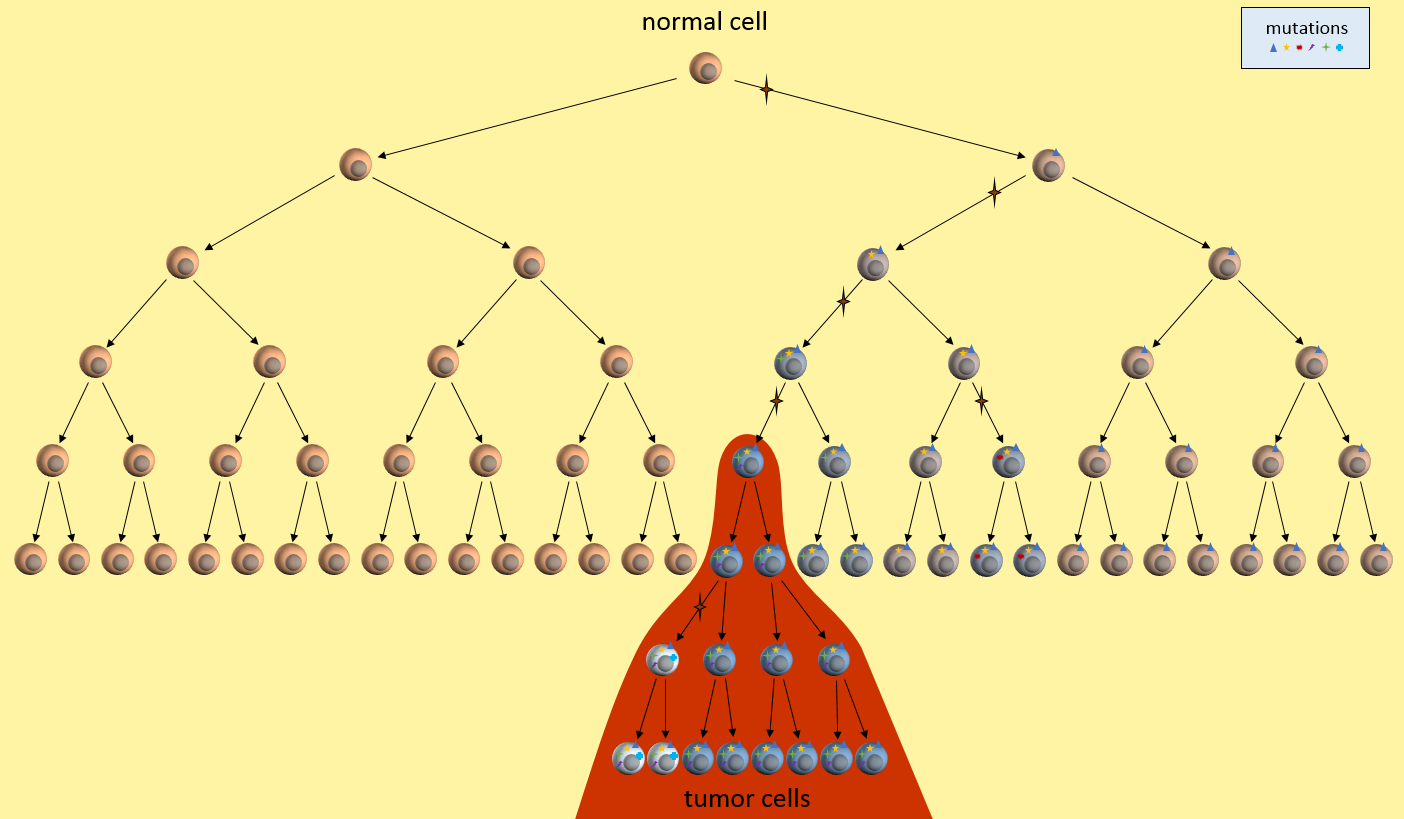
A cancer stem cell is very long-living, can accumulate genetic mutations over its lifetime, and then produce a nearly unlimited supply of cancer cells containing these mutations. Just as in the clonal evolution model, these cancer cells could continue to generate new mutations and divide uncontrollably.
[Whether CSC’s are actual adult stem cells that have become cancerous or are normal cells that have acquired “stem-like” properties is under active investigation.]
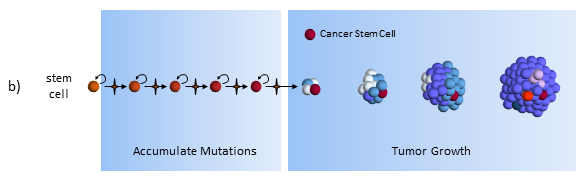
Tumor Growth under the Cancer Stem Cell Model
The key to the cancer stem cell model though, is the small colony of cancer stem cells (red cell above) that regenerate cancer cells but are killed differently than normal cancer cells.5 Killing off the stem cells will result in the eventual dissipation of the tumor as it can no longer regenerate.
Cancer Stem Cell Model on Chemotherapy
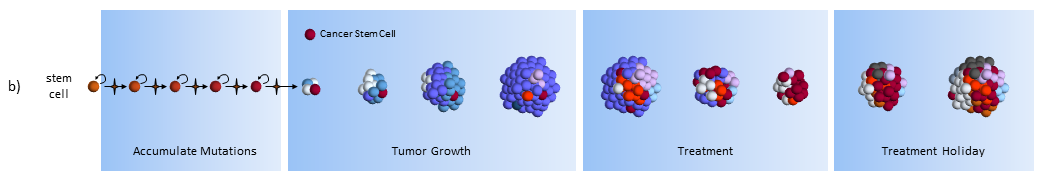
Cancer stem cell model response to chemotherapy treatments
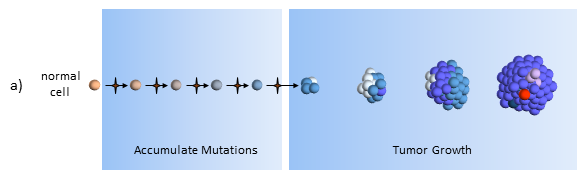
Accumulate Mutations
In the CSC model, a long-lived stem cell accumulates the cancer-causing mutations. It is believed that the property of a long lifetime allows it to accumulate all these mutations. A normal cell, with its much shorter lifespan, would be unlikely to accumulate enough mutations. The key difference is that at the core of the tumor is a CSC, sometimes called a tumor-initiating cell.
[I’m couching the discussion with phrases above like “it is believed”, but if you read the stem cell theory papers, these expressions of doubt are not presented. Some stem cell theorists say that a normal cell gains mutations and becomes stem cell-like to drive the cancer.]
Tumor Growth
The CSC is the major producer of all the new cancer cells3. The normal cancer cells (NCS) have limited cell division capability (just like normal cells). The CSC can continue to mutate which also results in tumor heterogeneity.
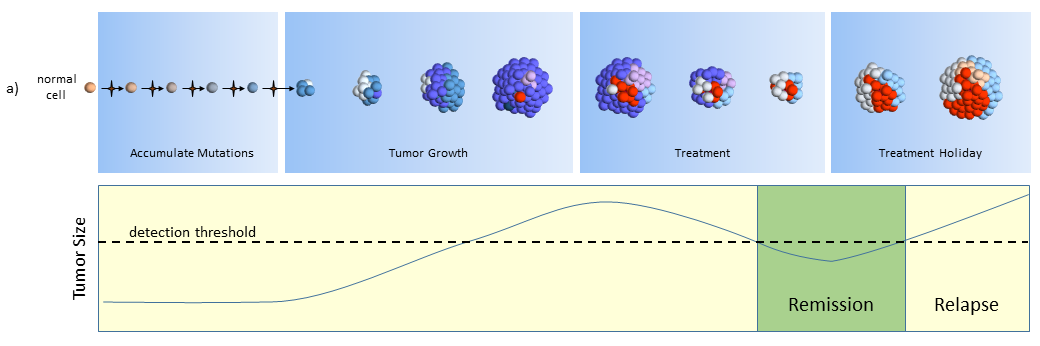
Treatment
As treatment begins, susceptible cells are again destroyed. However, CSC’s, which are slowly dividing, are not susceptible to chemotherapy3,5. To make matters worse, the CSC’s, acting like stem cells, see the tumor’s tissue damage and do what all stem cells do – regenerate new tissue (more CSC’s!). The result could again be a smaller tumor, but with an even larger concentration of CSC’s. In this model, what looks like good news on a CT scan (a smaller tumor) is really bad for the future.
[From a patient perspective, the CSC and clonal evolution model behave the same on scans, but the resulting tumor is very different. If the CSC model is correct, then in the long run, giving treatments that don’t kill the CSC’s is a bad thing to do.]
Treatment Holiday
After treatment, driven by many more CSC’s, the tumor growth accelerates.
[Question for Researchers: How does this square up with patients who have long-lasting remissions to chemotherapy?]
Cancer Stem Cell Model on Chemotherapy and Stem Cell Therapy
Under the CSC theory, the correct treatment protocol is to target the CSC’s themselves. Forget about the other cancer cells. Once the CSC’s are gone, the normal cancer cells cannot keep going by themselves and eventually perish. You can use a normal chemotherapy agent in addition to the CSC treatment in order to hasten the demise.
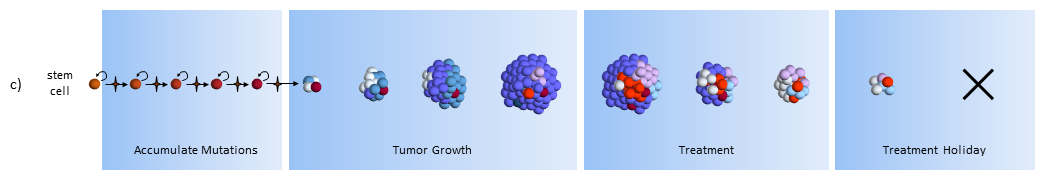
Cancer stem cell model response to chemotherapy and stem cell treatments
Treatment
As treatment begins, susceptible cells are again destroyed. In theory, stem cell therapy eliminates the CSC’s. Careful targeting of the CSC’s must be done to make sure that normal stem cells are not affected – which would probably be devastating to the patient.
Treatment Holiday
After treatment, with no CSC’s to replenish them, the normal cancer cells eventually die off.5
Controversy
The cancer stem cell model is not embraced by many cancer experts. The primary evidence is based on a set of experiments that break a tumor down and separate the tumor cells into different types. When 20,000 (or so) tumor cells of one type are transplanted into mice, a tumor does not take hold. When just 200 tumor cells of another type are transplanted, the tumor grows. This second set of tumor cells are considered CSC’s because they initiated human cancer growth in the mice. Experiments like these identify tumor cells with stem-like properties.
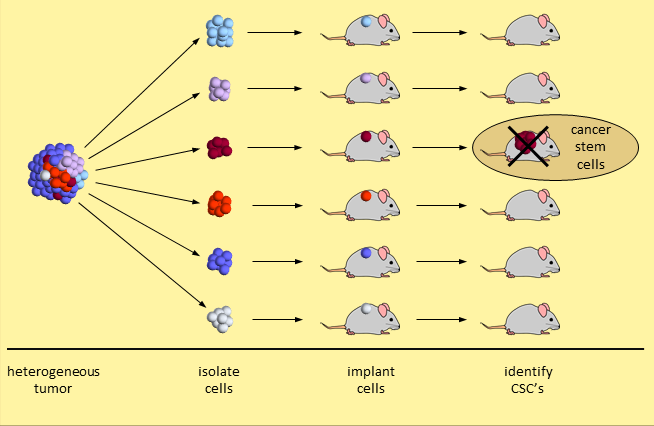
Identifying Cancer Stem Cells
Skeptical researchers say that a demonstration of human tumor cell growth in immunodeficient mice is insufficient. Growing human cancer cells in a mouse is too dissimilar an environment to provide proof that these are cancer stem cells.5
It should also be noted that testicular cancer that is curable with chemotherapy alone. If cancer stem cells were involved, this would not be possible. So apparently CSC’s do not drive all cancers.
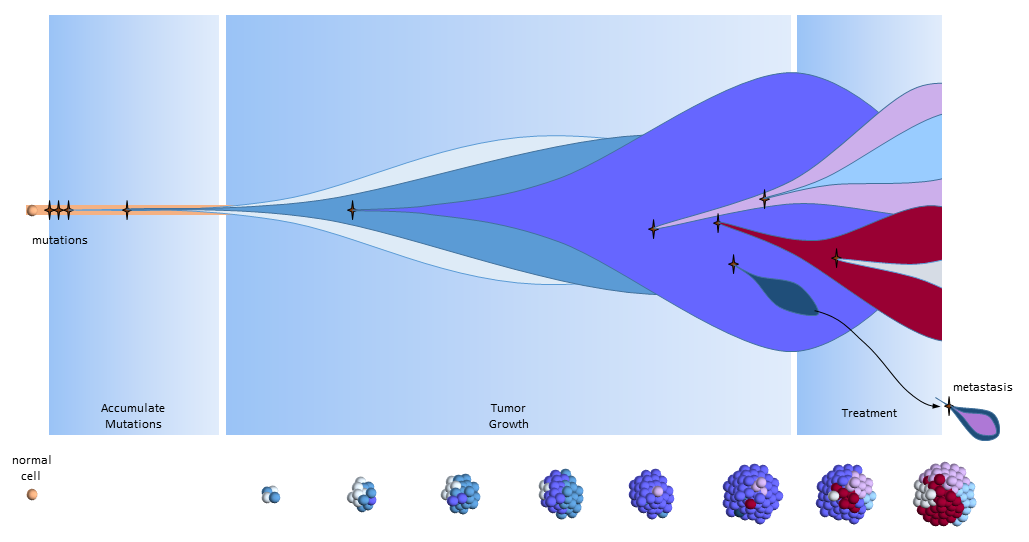
Another View of the Cancer Stem Cell Model
The figure below presents another way to look at tumor growth in the cancer stem call model. Each colored area represents a cell colony with a specific set of mutations. Time progresses to the right. The height of each colored area represents the quantity of cells in the colony. New mutations are represented by stars and may originate from any established colony. The figure shows that these new mutations only originate from the tumor’s cancer stem cells (dark red) and then compete for space and resources with other colonies.
Chemotherapy treatment is effective on the normal cancer cells, but has the opposite effect on cancer stem cells which multiply in response to the tissue damage. After treatment ends, a larger number of cancer stem cells are present to begin tumor regrowth.
Eventually, one of the colonies acquires the ability to metastasize and migrates to another organ.
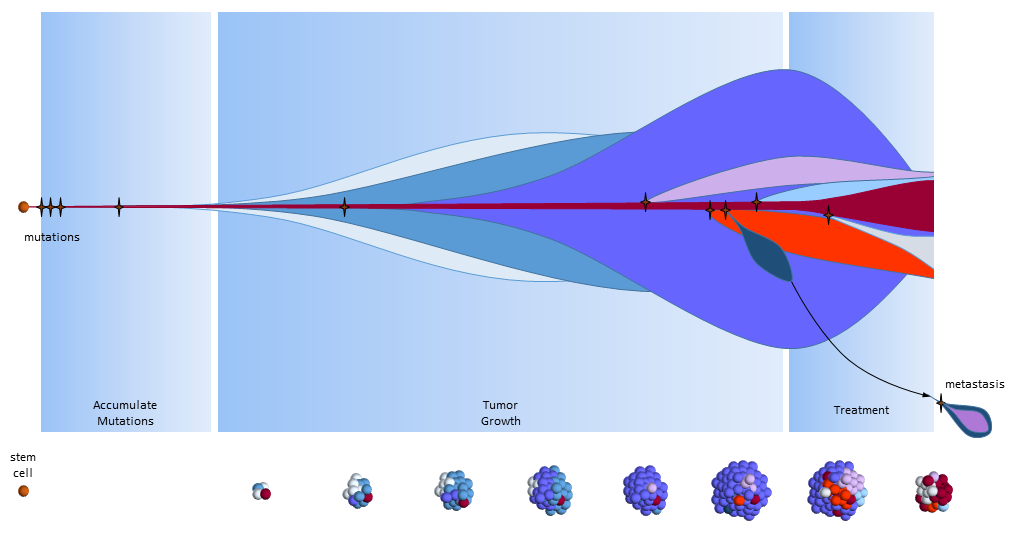
Cancer Stem Cell Model with Effective Chemotherapy
Summary
The cancer stem cell model is an alternate explanation for tumor growth and response to treatments. It was devised to try to explain failures of treatments based on the correctness of the clonal evolution model. The correctness of the cancer stem cell model is hotly debated.
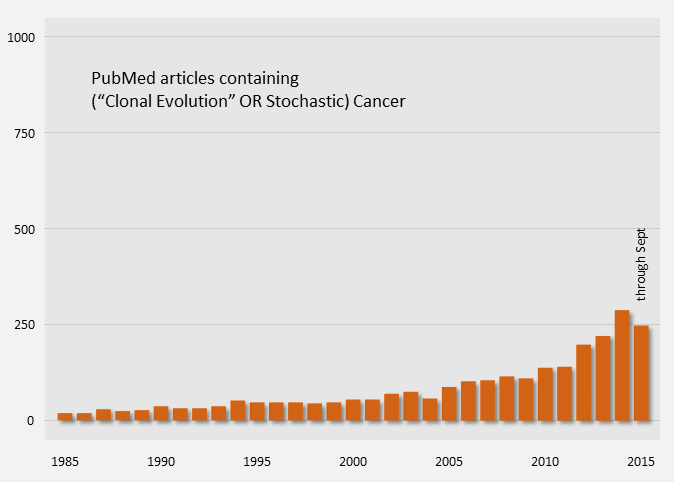
Publication counts of Cancer Stem Cell papers started in 2004 and have been on a steep rise, indicating that it is an active topic. For comparison, publication counts for the clonal evolution model are shown in orange below.
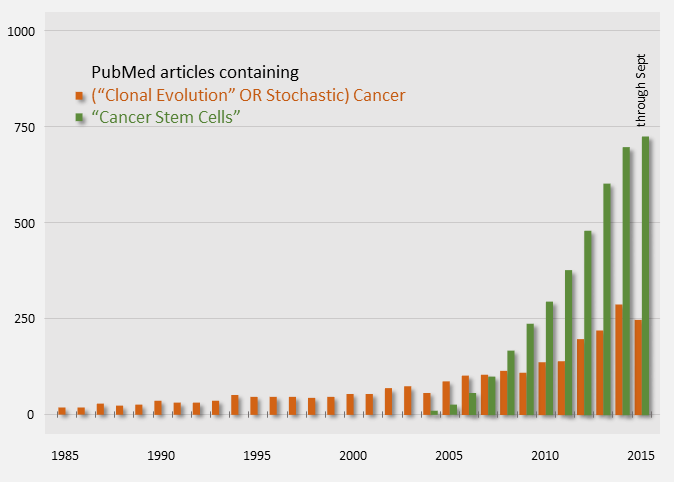
Cancer Model Publication Counts (PubMed)
Assuming that the cancer stem cell model is true, here are some points to consider.
- CSC’s are rare, “immortal” cells found in some cancers
- CSC’s are a tiny fraction of the total tumor (<1%)
- Radiotherapy and chemotherapy kill off normal cancer cells, but CSC’s respond by multiplying
- When normal cancer cells are depleted, CSC’s regenerate new cancer cells3,6
- The only cure is complete elimination of the CSC’s:
Surgical resection of solid tumors (before metastases) remains the only curative treatment common to both models.
References
[1] Nowell PC (October 1976). “The clonal evolution of tumor cell populations”. Nature 194(4260):23-8. PMID: 959840.
[2] Soltysova A, Altanerova V, et al. (2005). “Cancer stem cells”. Neoplasma 52(6):435-40. PMID: 16284686.
[3] University of Michigan Cancer Stem Cell Research Introduction web page (accessed 21 Sep 2015) http://www.mcancer.org/research/stem-cells/introduction
[4] University of Michigan Cancer Stem Cell Research Treatment web page (accessed 21 Sep 2015) http://www.mcancer.org/research/stem-cells/introduction/treatment-options
[5] EuroStemCell web page: Cancer: a disease of stem cells? (accessed 21 Sep 2015) http://www.eurostemcell.org/factsheet/cancer-disease-stem-cells
[6] Science in School web page: Cancer stem cells – hope for the future? (accessed 21 Sep 2015) http://www.scienceinschool.org/2011/issue21/cscs